Digital Case Study – Three
Case history
32 year old man presented with a 6 month history of abdominal pain, diahorrea and rectal bleeding. Colonoscopic examination identified multiple areas of erythema in the caecum and ascending colon, which were biopsied and submitted for histopathology.
Biopsy figures
- Figures 1, 2 and 3 – colon.
- Figures 4 and 5 – distribution and cytomorphology of abnormal cells within mucosa.
- Figures 6 and 7 – CD117 immunostain

Figure 1 – colon


Figure 3 – colon


Figure 5 – abnormal cells in mucosa

Figure 6 – CD117 IHC

Mastocytosis is a clonal neoplastic proliferation of mast cells that can accumulate in any organ system. The current WHO classification system subtypes mastocytosis into:
- Cutaneous mastocytosis
- Indolent systemic mastocytosis
- Systemic Mastocytosis (SM) with an associated clonal haematological non-mast cell lineage disease
- Aggressive systemic mastocytosis
- Mast cell leukaemia
- Mast cell sarcoma
- Extracutaneous mastocytoma [1].
The diagnosis of SM can be made when major criteria and one minor criterion or at least three minor criteria are present [1]. The major and minor criteria are as follows:
Major criteria: multifocal dense infiltrates of mast cells (>15 mast cells in aggregates) detected in sections of bone marrow and/or other extracutaneous organ(s).
Minor criteria:
- In biopsy sections of bone marrow or other extracutaneous organs, >25% of the mast cells in the infiltrate are spindle-shaped or have atypical morphology or, of all mast cells in bone marrow aspirate smears, >25% are immature or atypical.
- Detection of an activating point mutation at codon 816 of KIT in bone marrow, blood or another extracutaneous organ.
- Mast cells in bone marrow, blood or other extracutaneous organs express CD2 and/or CD25 in addition to normal mast cell markers.
- Serum total tryptase persistently exceeds 20 ng/ml (unless there is an associated clonal myeloid disorder, in which case this parameter is not valid).
The presence of atypical mast cell infiltrates within gastrointestinal (GI) mucosal biopsies can be used to define SM according to WHO criteria and may be the first diagnosis in up to 67% cases [2]. Gastrointestinal involvement is common, with up to 92% of patients describing bloating, reflux, nausea, vomiting, abdominal pain, diarrhoea and weight loss [2-6]. These symptoms are thought to be due to direct infiltration of the mucosa by mast cells resulting in malabsorption and release of mast cell mediators such as histamines and leukotrienes.
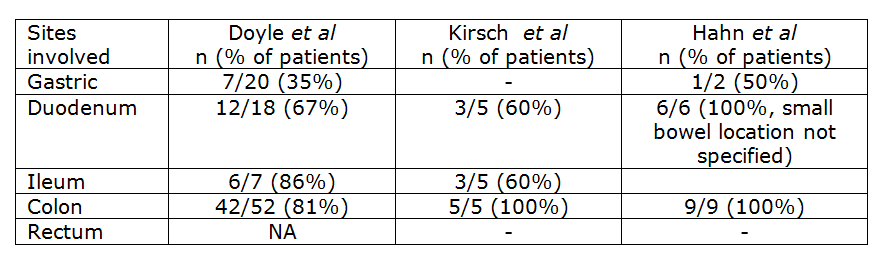
Although GI involvement is common in SM it is not necessarily a straight forward diagnosis as the histological changes and clinical presentation can mimic other disorders such as eosinophilic gastroenteritis and inflammatory bowel disease [4]. Endoscopic findings can also be subtle with 48% of patients described as having no abnormality in the study by Doyle et al [2] Documented endoscopic changes include erythema, congestion, erosions, loss of mucosal folds, granularity and nodularity [2,4,5]. Biopsies taken from abnormal mucosa are much more likely to show involvement by mastocytosis (93% of cases), however up to 63% of biopsies taken from apparently normal mucosa can also show the abnormal mast cell infiltrate [2]. Involvement of the gastrointestinal tract can also be patchy, therefore taking multiple biopsies from different sites and even apparently normal mucosa is optimal for diagnosis [6]. The stomach appears to be less commonly involved than small and large bowel as documented in the table below.